
BAVENCIO 20 mg-mL solution à diluer pour perfusion, boîte de 1 flacon de 10 ml
Retiré du marché le : 01/03/2021
Dernière révision : 07/07/2020
Taux de TVA : 0%
Laboratoire exploitant : MERCK EUROPE B.V.
Source :

Bavencio est indiqué en monothérapie dans le traitement d'entretien de première ligne des patients adultes atteints d'un carcinome urothélial (CU) localement avancé ou métastatique, dont la maladie n'a pas progressé après une chimiothérapie d'induction de première ligne à base de sels de platine et présentant un score ECOG 0 ou 1.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Réactions liées à la perfusion
Des réactions liées à la perfusion, pouvant être sévères, ont été signalées chez des patients traités par avélumab (voir rubrique Effets indésirables).
Les patients doivent être surveillés afin de détecter des signes et symptômes de réactions liées à la perfusion, notamment fièvre, frissons, bouffées vasomotrices, hypotension, dyspnée, sifflement respiratoire, dorsalgie, douleur abdominale et urticaire.
En cas de réaction liée à la perfusion de grade 3 ou de grade 4, la perfusion doit être interrompue et le traitement par avélumab doit être définitivement arrêté (voir rubrique Posologie et mode d'administration).
Pour les réactions liées à la perfusion de grade 1, le débit de perfusion doit être réduit de 50 % pour la perfusion en cours. En cas de réaction liée à la perfusion de grade 2, la perfusion doit être provisoirement arrêtée jusqu'au retour à un grade 1 ou jusqu'à la disparition de la réaction, et elle sera reprise ensuite à un débit de perfusion réduit de 50 % (voir rubrique Posologie et mode d'administration).
En cas de réapparition d'une réaction liée à la perfusion de grade 1 ou de grade 2, le patient pourra continuer à recevoir avélumab sous étroite surveillance, après ajustement adéquat du débit de perfusion et administration d'une prémédication par paracétamol et antihistaminique (voir rubrique Posologie et mode d'administration).
Chez 98,6 % (433/439) des patients ayant présenté des réactions liées à la perfusion au cours des essais cliniques, la première réaction s'est produite lors des 4 premières perfusions dont 2,7 % (12/439) étaient de grade ≥ 3. Chez les 1,4 % (6/439) de patients restants, les réactions liées à la perfusion se sont produites après les 4 premières perfusions et étaient toutes de grade 1 ou de grade 2.
Effets indésirables d'origine immunologique
La plupart des effets indésirables d'origine immunologique survenus avec avélumab ont été réversibles et ont pu être gérés au moyen d'interruptions temporaires ou définitives du traitement par avélumab, de l'administration de corticoïdes et/ou de soins de support.
Lorsqu'un effet indésirable d'origine immunologique est suspecté, une évaluation adéquate doit être réalisée afin de confirmer l'étiologie ou d'exclure les autres causes. Selon la sévérité de l'effet indésirable, le traitement par avélumab devra être suspendu et une corticothérapie administrée. Si des corticoïdes sont utilisés pour traiter un effet indésirable, une réduction progressive de leurs doses doit être initiée sur une période d'au moins 1 mois à partir de l'amélioration.
Lorsque les effets indésirables d'origine immunologique n'ont pas pu être contrôlés par la corticothérapie, l'administration d'autres immunosuppresseurs systémiques pourra être envisagée.
Pneumopathie d'origine immunologique
Des cas de pneumopathie d'origine immunologique ont été observés chez des patients traités par avélumab. Un cas avec une issue fatale a été signalé chez des patients traités par avélumab (voir rubrique Effets indésirables).
L'apparition de signes et symptômes de pneumopathie d'origine immunologique doit être surveillée chez les patients et les causes de pneumopathie autres qu'une origine immunologique doivent être exclues. En cas de suspicion, la pneumopathie doit être confirmée par un examen radiographique.
Une corticothérapie doit être administrée en cas d'événement de grade ≥ 2 (dose initiale de 1 à 2 mg/kg/jour de prednisone ou équivalent, suivie d'une réduction progressive du corticoïde).
Le traitement par avélumab doit être suspendu jusqu'à disparition de la réaction en cas de pneumopathie d'origine immunologique de grade 2 et définitivement arrêté en cas de pneumopathie d'origine immunologique de grade 3, de grade 4, ou de grade 2 si elle est récurrente (voir rubrique Posologie et mode d'administration).
Hépatite d'origine immunologique
Des cas d'hépatite d'origine immunologique ont été observés chez des patients traités par avélumab. Deux cas avec une issue fatale ont été signalés chez des patients traités par avélumab (voir rubrique Effets indésirables).
Les altérations de la fonction hépatique et les symptômes d'hépatite d'origine immunologique doivent être surveillés chez les patients et les causes d'hépatite autres que l'origine immunologique doivent être exclues.
Une corticothérapie doit être administrée en cas d'événements de grade ≥ 2 (dose initiale de 1 à 2 mg/kg/jour de prednisone ou équivalent, suivie d'une réduction progressive du corticoïde).
Le traitement par avélumab doit être suspendu jusqu'à disparition de la réaction en cas d'hépatite d'origine immunologique de grade 2 et définitivement arrêté en cas d'hépatite d'origine immunologique de grade 3 ou de grade 4 (voir rubrique Posologie et mode d'administration).
Colite d'origine immunologique
Des cas de colite d'origine immunologique ont été signalés chez des patients traités par avélumab (voir rubrique Effets indésirables).
L'apparition de signes et symptômes de colite d'origine immunologique doit être surveillée chez les patients et les causes de colite autres qu'une origine immunologique doivent être exclues. Une corticothérapie doit être administrée en cas d'événements de grade ≥ 2 (dose initiale de 1 à 2 mg/kg/jour de prednisone ou équivalent, suivie d'une réduction progressive du corticoïde).
Le traitement par avélumab doit être suspendu jusqu'à disparition de la réaction en cas de colite d'origine immunologique de grade 2 ou de grade 3 et définitivement arrêté en cas de colite d'origine immunologique de grade 4, ou de grade 3 si elle est récurrente (voir rubrique Posologie et mode d'administration).
Pancréatite d'origine immunologique
Des cas de pancréatite d'origine immunologique ont été signalés chez des patients traités par avélumab. Deux cas avec une issue fatale ont été signalés chez des patients traités par avélumab en association avec axitinib (voir rubrique Effets indésirables).
L'apparition de signes et symptômes de pancréatite d'origine immunologique doit être surveillée chez les patients. Chez les patients symptomatiques, une consultation en gastroentérologie et des examens de laboratoire (y compris des examens d'imagerie médicale) sont indiqués pour assurer l'instauration précoce de mesures appropriées. Une corticothérapie doit être administrée en cas de pancréatite d'origine immunologique (dose initiale de 1 à 2 mg/kg/jour de prednisone ou équivalent, suivie d'une réduction progressive du corticoïde).
Le traitement par avélumab doit être suspendu en cas de suspicion de pancréatite d'origine immunologique et définitivement arrêté en cas de confirmation de la pancréatite d'origine immunologique (voir rubrique Posologie et mode d'administration).
Myocardite d'origine immunologique
Des cas de myocardite d'origine immunologique ont été signalés chez des patients traités par avélumab. Deux cas avec une issue fatale ont été signalés chez des patients traités par avélumab en association avec axitinib (voir rubrique Effets indésirables).
L'apparition de signes et symptômes de myocardite d'origine immunologique doit être surveillée chez les patients. Chez les patients symptomatiques, une consultation en cardiologie et des examens de laboratoire sont indiqués pour assurer l'instauration précoce des mesures appropriées. Une corticothérapie doit être administrée en cas de myocardite d'origine immunologique (dose initiale de 1 à 2 mg/kg/jour de prednisone ou équivalent, suivie d'une réduction progressive du corticoïde). En l'absence d'amélioration après 24 heures de corticothérapie, une immunosuppression supplémentaire (par ex. par mycophénolate, infliximab ou globuline anti-thymocyte) doit être envisagée.
Le traitement par avélumab doit être suspendu en cas de suspicion de myocardite d'origine immunologique et définitivement arrêté en cas de confirmation de la myocardite d'origine immunologique (voir rubrique Posologie et mode d'administration).
Endocrinopathies d'origine immunologique
Des cas de troubles thyroïdiens d'origine immunologique, d'insuffisance surrénalienne d'origine immunologique et de diabète de type 1 ont été rapportés chez des patients traités par avélumab (voir rubrique Effets indésirables). Les patients doivent être surveillés afin de détecter des signes et symptômes cliniques d'endocrinopathies. Le traitement par avélumab doit être suspendu jusqu'à disparition de la réaction en cas d'endocrinopathies de grade 3 ou de grade 4 (voir rubrique Posologie et mode d'administration).
Troubles thyroïdiens (hypothyroïdie/hyperthyroïdie)
Des troubles thyroïdiens peuvent survenir à tout moment pendant le traitement (voir rubrique Effets indésirables).
Les altérations de la fonction thyroïdienne doivent être surveillées chez les patients (en début de traitement, régulièrement au cours du traitement et dès lors qu'un suivi est indiqué en fonction de l'évaluation clinique), ainsi que les signes cliniques et les symptômes de troubles thyroïdiens. L'hypothyroïdie doit être prise en charge au moyen d'un traitement hormonal substitutif et l'hyperthyroïdie, au moyen d'un médicament antithyroïdien, selon les besoins.
Le traitement par avélumab doit être suspendu en cas de troubles thyroïdiens de grade 3 ou de grade 4 (voir rubrique Posologie et mode d'administration).
Insuffisance surrénalienne
L'apparition de signes et symptômes d'insuffisance surrénalienne doit être surveillée chez les patients pendant et après le traitement. Une corticothérapie doit être administrée (1 à 2 mg/kg/jour de prednisone par voie intraveineuse ou équivalent oral) en cas d'insuffisance surrénalienne de grade ≥ 3, suivie d'une réduction progressive de la dose jusqu'à ≤ 10 mg/jour.
Le traitement par avélumab doit être suspendu en cas d'insuffisance surrénalienne symptomatique de grade 3 ou de grade 4 (voir rubrique Posologie et mode d'administration).
Diabète de type 1
Avélumab peut provoquer un diabète de type 1, y compris une acidocétose diabétique (voir rubrique Effets indésirables).
Les patients doivent être surveillés pour rechercher une hyperglycémie ou d'autres signes et symptômes du diabète. Un traitement par insuline doit être instauré pour le diabète de type 1. En cas d'hyperglycémie de grade ≥ 3, le traitement par avélumab doit être suspendu et des anti- hyperglycémiants doivent être administrés. Le traitement par avélumab pourra être repris après obtention du contrôle métabolique sous insulinothérapie.
Néphrite et dysfonction rénale d'origine immunologique
Avélumab peut provoquer une néphrite d'origine immunologique (voir rubrique Effets indésirables).
Les patients doivent être surveillés pour rechercher des taux sériques élevés de créatinine avant le traitement et, à intervalles réguliers, pendant le traitement. En cas de néphrite de grade ≥ 2, des corticoïdes doivent être administrés (dose initiale de 1 à 2 mg/kg/jour de prednisone ou équivalent, puis diminution progressive). En cas de néphrite de grade 2 ou de grade 3, le traitement par avélumab doit être suspendu jusqu'au retour à un grade ≤ 1. En cas de néphrite de grade 4, le traitement par avélumab doit être arrêté définitivement.
Autres effets indésirables d'origine immunologique
D'autres effets indésirables d'origine immunologique cliniquement importants ont été rapportés chez moins de 1 % des patients : myosite, hypopituitarisme, uvéite et syndrome de Guillain-Barré (voir rubrique Effets indésirables).
Lorsqu'un effet indésirable d'origine immunologique est suspecté, une évaluation adéquate doit être réalisée afin de confirmer l'étiologie ou d'exclure d'autres causes. Selon la sévérité de l'effet indésirable, le traitement par avélumab devra être suspendu et une corticothérapie devra être administrée. Le traitement par avélumab devra être repris une fois que l'effet indésirable d'origine immunologique sera revenu à un grade 1 ou inférieur, après réduction progressive du corticoïde. Le traitement par avélumab doit être définitivement arrêté en cas de récidive d'un effet indésirable d'origine immunologique de grade 3 et en cas d'effet indésirable d'origine immunologique de grade 4 (voir rubrique Posologie et mode d'administration).
Hépatotoxicité (en association avec axitinib)
Des cas d'hépatotoxicité ont été signalés chez des patients traités par avélumab en association avec axitinib et la fréquence d'élévation des taux d'ALAT ou d'ASAT de grade 3 et de grade 4 était plus élevée que celle observée avec avélumab seul (voir rubrique Effets indésirables).
Les modifications de la fonction hépatique et les symptômes hépatiques doivent être surveillés plus fréquemment chez les patients traités par avélumab en association avec axitinib que chez les patients traités par avélumab en monothérapie.
Le traitement par avélumab doit être suspendu en cas d'hépatotoxicité de grade 2 jusqu'à résolution de la toxicité et définitivement arrêté en cas d'hépatotoxicité de grade 3 ou de grade 4. Une corticothérapie doit être envisagée en cas d'évènements de grade ≥ 2.
Patients exclus des études cliniques
Les patients atteints des affections suivantes ont été exclus des essais cliniques : métastases actives du système nerveux central (SNC), maladie auto-immune active ou antérieure, antécédents d'autres tumeurs malignes au cours des 5 années précédentes, greffe d'organe, affection nécessitant un traitement immunosuppresseur ou infection active par le VIH, ou hépatite B ou C.
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement « sans sodium ».
Avélumab est associé à des effets indésirables d'origine immunologique. La plupart d'entre eux, y compris les réactions sévères, se sont résolus après instauration d'un traitement médical approprié ou arrêt d'avélumab (voir « Description de certains effets indésirables particuliers » ci-dessous).
Résumé du profil de sécurité
La sécurité d'emploi d'avélumab en monothérapie a été évaluée chez 1 738 patients atteints de tumeurs solides, y compris de carcinome à cellules de Merkel (CCM) métastatique, ayant reçu 10 mg/kg d'avélumab toutes les 2 semaines dans des études cliniques. Dans cette population de patients, les effets indésirables les plus fréquents avec avélumab étaient des cas de fatigue (32,4 %), des nausées (25,1 %), des diarrhées (18,9 %), une diminution de l'appétit (18,4 %), une constipation (18,4 %), des réactions liées à la perfusion (17,1 %), une perte de poids (16,6 %) et des vomissements (16,2 %).
Les effets indésirables de grade ≥ 3 les plus fréquents étaient une anémie (6,0 %), une dyspnée (3,9 %) et des douleurs abdominales (3,0 %). Les effets indésirables graves étaient des effets indésirables d'origine immunologique et des réactions liées à la perfusion (voir rubrique Mises en garde et précautions d'emploi).
Tableau récapitulatif des effets indésirables
Les effets indésirables rapportés avec avélumab en monothérapie chez les patients atteints de tumeurs solides telles que le CCM métastatique et CU localement avancé ou métastatique sont présentés dans le Tableau 2. Dans ces études, avélumab était administré à la dose de 10 mg/kg toutes les 2 semaines.
Ces réactions sont présentées par classe de système d'organe et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000). Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 2 : Effets indésirables survenus chez les patients traités par avélumab en monothérapie
| Fréquence | Effets indésirables |
| Infections et infestations | |
| Très fréquent | Infection urinaire |
| Affections hématologiques et du système lymphatique | |
| Très fréquent | Anémie |
| Fréquent | Lymphopénie |
| Peu fréquent | Thrombocytopénie, éosinophilie§ |
| Affections du système immunitaire | |
| Peu fréquent | Hypersensibilité médicamenteuse, réaction anaphylactique d'hypersensibilité, hypersensibilité de type I |
| Affections endocriniennes | |
| Fréquent | Hypothyroïdie* |
| Peu fréquent | Insuffisance surrénalienne*, hyperthyroïdie*, thyroïdite*, thyroïdite auto-immune*, insuffisance corticosurrénalienne aiguë*, hypothyroïdie auto-immune*, hypopituitarisme* |
| Troubles du métabolisme et de la nutrition | |
| Très fréquent | Diminution de l'appétit |
| Fréquence | Effets indésirables |
| Peu fréquent | Diabète*, diabète de type I*, hyperglycémie* |
| Affections du système nerveux | |
| Fréquent | Céphalée, vertiges, neuropathie périphérique |
| Peu fréquent | Syndrome de Guillain-Barré*, syndrome de Miller-Fisher* |
| Affections oculaires | |
| Peu fréquent | Uvéite* |
| Affections cardiaques | |
| Rare | Myocardite* |
| Affections vasculaires | |
| Fréquent | Hypertension, hypotension |
| Peu fréquent | Bouffées vasomotrices |
| Affections respiratoires, thoraciques et médiastinales | |
| Très fréquent | Toux, dyspnée |
| Fréquent | Pneumopathie* |
| Peu fréquent | Pneumopathie interstitielle* |
| Affections gastro-intestinales | |
| Très fréquent | Nausées, diarrhée, constipation, vomissements, douleur abdominale |
| Fréquent | Sécheresse buccale |
| Peu fréquent | Colite*, colite auto-immune*, entérocolite*, iléus, pancréatite auto-immune*, entérite*, proctite* |
| Rare | Pancréatite* |
| Affections hépatobiliaires | |
| Peu fréquent | Hépatite auto-immune*, insuffisance hépatique aiguë*, insuffisance hépatique*, hépatite*, hépatotoxicité* |
| Affections de la peau et du tissu sous-cutané | |
| Fréquent | Éruption cutanée*, prurit*, éruption maculo-papuleuse*, sécheresse de la peau |
| Peu fréquent | Éruption prurigineuse*, érythème*, éruption généralisée*, psoriasis*, éruption érythémateuse*, éruption maculaire*, éruption papuleuse*, dermatite exfoliative*, érythème polymorphe*, pemphigoïde*, prurit généralisé*, eczéma, dermatite, vitiligo*, purpura*, dermatite psoriasiforme*, éruption médicamenteuse*, lichen plan* |
| Affections musculo-squelettiques et systémiques | |
| Très fréquent | Dorsalgie, arthralgie |
| Fréquent | Myalgie |
| Peu fréquent | Myosite*, arthrite*, polyarthrite*, oligoarthrite*, polyarthrite rhumatoïde* |
| Affections du rein et des voies urinaires | |
| Peu fréquent | Néphrite tubulo-interstitielle*, insuffisance rénale*, néphrite* |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent | Fatigue, fièvre, oedème périphérique |
| Fréquent | Asthénie, frissons, syndrome pseudo-grippal |
| Peu fréquent | Syndrome de réponse inflammatoire généralisée* |
| Investigations | |
| Très fréquent | Perte de poids |
| Fréquent | Élévation des taux de gamma-glutamyltransférase, élévation de la phosphatase alcaline plasmatique, élévation de l'amylase, élévation de la lipase, élévation de la créatinine plasmatique |
| Peu fréquent | Élévation des taux d'alanine aminotransférase (ALAT)*, élévation des taux d'aspartate aminotransférase (ASAT)*, élévation de la créatine phosphokinase plasmatique* et élévation des taux de transaminases plasmatiques*, élévation de la thyréostimuline (TSH) plasmatique*, diminution de la thyroxine libre* |
| Lésions, intoxications et complications liées aux procédures | |
| Très fréquent | Réaction liée à la perfusion |
* Effet indésirable d'origine immunologique d'après la revue médicale
§ Réaction uniquement observée dans l'étude EMR100070-003 (Partie B) après la date limite de recueil des données, il s'agit donc d'une estimation de la fréquence
Carcinome à cellules rénales (CCR)
Résumé du profil de sécurité
La sécurité d'emploi d'avélumab en association avec axitinib a été évaluée chez 489 patients atteints de CCR à un stade avancé ayant reçu 10 mg/kg d'avélumab toutes les 2 semaines et 5 mg d'axitinib par voie orale deux fois par jour dans deux études cliniques.
Dans cette population de patients, les effets indésirables les plus fréquents étaient la diarrhée (62,8 %), l'hypertension (49,3 %), la fatigue (42,9 %), les nausées (33,5 %), la dysphonie (32,7 %), la diminution de l'appétit (26,0 %), l'hypothyroïdie (25,2 %), la toux (23,7 %), les céphalées (21,3 %), la dyspnée (20,9 %) et lesarthralgies (20,9 %).
Tableau récapitulatif des effets indésirables
Les effets indésirables rapportés chez les 489 patients atteints de CCR à un stade avancé et traités par avélumab en association avec axitinib au cours de deux études cliniques sont présentés dans le Tableau 3.
Ces réactions sont présentées par classe de système d'organe et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000). Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 3 : Effets indésirables survenus chez les patients traités par avélumab en association avec axitinib au cours des études cliniques B9991002 et B9991003
| Fréquence | Effets indésirables |
| Infections et infestations | |
| Peu fréquent | Eruption pustuleuse |
| Affections hématologiques et du système lymphatique | |
| Fréquent | Anémie, thrombocytopénie |
| Peu fréquent | Lymphopénie, éosinophilie |
| Affections du système immunitaire | |
| Fréquent | Hypersensibilité |
| Affections endocriniennes | |
| Très fréquent | Hypothyroïdie |
| Fréquent | Hyperthyroïdie, insuffisance surrénalienne, thyroïdite |
| Peu fréquent | Thyroïdite auto-immune, hypophysite |
| Troubles du métabolisme et de la nutrition | |
| Très fréquent | Diminution de l'appétit |
| Fréquent | Hyperglycémie |
| Peu fréquent | Diabète, diabète de type I |
| Affections du système nerveux | |
| Très fréquent | Céphalée, vertiges |
| Fréquent | Neuropathie périphérique |
| Affections cardiaques | |
| Peu fréquent | Myocardite |
| Affections vasculaires | |
| Très fréquent | Hypertension |
| Fréquent | Hypotension, bouffées vasomotrices |
| Affections respiratoires, thoraciques et médiastinales | |
| Très fréquent | Dysphonie, toux, dyspnée |
| Fréquent | Pneumopathie |
| Affections gastro-intestinales | |
| Très fréquent | Diarrhée, nausées, constipation, vomissements, douleur abdominale |
| Fréquent | Sécheresse buccale, colite |
| Peu fréquent | Colite auto-immune, pancréatite auto-immune, entérocolite, iléus, pancréatite nécrosante |
| Affections hépatobiliaires | |
| Fréquent | Fonction hépatique anormale |
| Peu fréquent | Hépatite, hépatotoxicité, hépatite à médiation immunitaire, trouble hépatique |
| Fréquence | Effets indésirables |
| Affections de la peau et du tissu sous-cutané | |
| Très fréquent | Éruption cutanée, prurit |
| Fréquent | Éruption prurigineuse, éruption maculo-papuleuse, prurit généralisé, dermatite acnéiforme, érythème, éruption maculaire, éruption papuleuse, éruption érythémateuse, dermatite, eczéma, éruption généralisée |
| Peu fréquent | Éruption d'origine médicamenteuse, érythème polymorphe, psoriasis |
| Affections musculo-squelettiques et systémiques | |
| Très fréquent | Arthralgie, dorsalgie, myalgie |
| Affections du rein et des voies urinaires | |
| Fréquent | Insuffisance rénale aiguë |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent | Fatigue, frissons, asthénie, fièvre |
| Fréquent | Œdème périphérique, syndrome pseudo-grippal |
| Investigations | |
| Très fréquent | Perte de poids, élévation des taux d'alanine aminotransférase (ALAT), élévation des taux d'aspartate aminotransférase (ASAT) |
| Fréquent | Élévation de la créatinine plasmatique, élévation de l'amylase, élévation de la lipase, élévation de la gamma-glutamyltransférase, élévation de la phosphatase alcaline plasmatique, élévation de la créatine phosphokinase plasmatique, diminution de la thyréostimuline (TSH) plasmatique, élévation des taux de transaminases plasmatiques |
| Peu fréquent | Augmentation des paramètres biologiques du bilan hépatique |
| Lésions, intoxications et complications liées aux procédures | |
| Très fréquent | Réaction liée à la perfusion |
Description de certains effets indésirables particuliers
Les données relatives aux effets indésirables d'origine immunologique pour avélumab en monothérapie reposent sur les 1 650 patients atteints de tumeurs solides dans l'étude de phase I EMR100070-001 et les 88 patients dans l'étude EMR100070-003 et, pour l'avélumab en association avec axitinib, sur les 489 patients des études B9991002 et B9991003.
Les recommandations pour la prise en charge de ces effets indésirables sont décrites dans la rubrique Mises en garde et précautions d'emploi.
Pneumopathie d'origine immunologique
Chez les patients traités par avélumab en monothérapie, des pneumopathies d'origine immunologique ont été observées chez 1,2 % (21/1 738) des patients. L'issue a été fatale pour 1 de ces patients (0,1 %). La pneumopathie d'origine immunologique était de grade 4 chez 1 patient (0,1 %) et de grade 3 chez 5 patients (0,3 %).
Le délai médian d'apparition de la pneumopathie d'origine immunologique était de 2,5 mois (intervalle : 3 jours à 11 mois). La durée médiane de la réaction était de 7 semaines (intervalle : 4 jours à plus de 4 mois).
L'administration d'avélumab a été interrompue chez 0,3 % (6/1 738) des patients en raison d'une pneumopathie d'origine immunologique. Les 21 patients ayant présenté une pneumopathie d'origine immunologique ont tous été traités par corticothérapie et 17 (81 %) des 21 patients ont reçu une corticothérapie à forte dose pendant une durée médiane de 8 jours (intervalle : 1 jour à 2,3 mois). La pneumopathie d'origine immunologique était résolue chez 12 (57 %) des 21 patients à la date limite de recueil des données.
Chez les patients traités par avélumab en association avec axitinib, des pneumopathies d'origine immunologique ont été observées chez 0,6 % (3/489) des patients. Aucun d'entre eux n'a présenté une pneumopathie d'origine immunologique de grade ≥ 3.
Le délai médian d'apparition de la pneumopathie d'origine immunologique était de 3,7 mois (intervalle : 2,7 mois à 8,6 mois). La durée médiane de la réaction était de 2,6 mois (intervalle : 3,3 semaines à plus de 7,9 mois).
L'administration d'avélumab n'a été interrompue chez aucun patient en raison d'une pneumopathie d'origine immunologique. Les 3 patients ayant présenté une pneumopathie d'origine immunologique ont été traités par une corticothérapie à forte dose pendant une durée médiane de 3,3 mois (intervalle : 3 semaines à 22,3 mois). La pneumopathie d'origine immunologique était résolue chez 2 (66,7 %) des 3 patients à la date limite de recueil des données.
Hépatite d'origine immunologique
Chez les patients traités par avélumab en monothérapie, des hépatites d'origine immunologique ont été observées chez 0,9 % (16/1 738) des patients. L'issue a été fatale pour 2 de ces patients (0,1 %), et l'hépatite d'origine immunologique était de grade 3 chez 11 patients (0,6 %).
Le délai médian d'apparition de l'hépatite d'origine immunologique était de 3,2 mois (intervalle : 1 semaine à 15 mois). La durée médiane de la réaction était de 2,5 mois (intervalle : 1 jour à plus de 7,4 mois).
L'administration d'avélumab a été interrompue chez 0,5 % (9/1 738) des patients en raison d'une hépatite d'origine immunologique. Les 16 patients ayant présenté une hépatite d'origine immunologique ont tous été traités par corticothérapie et 15 (94 %) des 16 patients ont reçu une corticothérapie à forte dose pendant une durée médiane de 14 jours (intervalle : 1 jour à 2,5 mois). L'hépatite d'origine immunologique était résolue chez 9 (56 %) des 16 patients à la date limite de recueil des données.
Chez les patients traités par avélumab en association avec axitinib, des hépatites d'origine immunologique ont été observées chez 6,3 % (31/489) des patients. L'hépatite d'origine immunologique était de grade 3 chez 18 de ces patients (3,7 %) et de grade 4 chez 3 patients (0,6 %).
Le délai médian d'apparition de l'hépatite d'origine immunologique était de 2,3 mois (intervalle : 2,1 semaines à 14,5 mois). La durée médiane de la réaction était de 2,1 semaines (intervalle : 2 jours à 8,9 mois).
L'administration d'avélumab a été interrompue chez 4,7 % (23/489) des patients en raison d'une hépatite d'origine immunologique. Les 31 patients ayant présenté une hépatite d'origine immunologique ont tous été traités : 30 (96,8 %) patients ont reçu une corticothérapie et 1 patient a reçu un immunosuppresseur non stéroïdien. Vingt-huit (90,3 %) des 31 patients ont suivi une corticothérapie à forte dose pendant une durée médiane de 2,4 semaines (intervalle : 1 jour à 10,2 mois). L'hépatite d'origine immunologique était résolue chez 27 (87,1 %) des 31 patients à la date limite de recueil des données.
Colite d'origine immunologique
Chez les patients traités par avélumab en monothérapie, des colites d'origine immunologique ont été observées chez 1,5 % (26/1 738) des patients. Chez 7 de ces patients (0,4 %), la colite d'origine immunologique a été de grade 3.
Le délai médian d'apparition de la colite d'origine immunologique était de 2,1 mois (intervalle : 2 jours à 11 mois). La durée médiane de la réaction était de 6 semaines (intervalle : 1 jour à plus de 14 mois).
L'administration d'avélumab a été interrompue chez 0,5 % (9/1 738) des patients en raison d'une colite d'origine immunologique. Les 26 patients ayant présenté une colite d'origine immunologique ont tous été traités par corticothérapie et 15 (58 %) des 26 patients ont reçu une corticothérapie à forte dose pendant une durée médiane de 19 jours (intervalle : 1 jour à 2,3 mois). La colite d'origine immunologique était résolue chez 18 (70 %) des 26 patients à la date limite de recueil des données.
Chez les patients traités par avélumab en association avec axitinib, des colites d'origine immunologique ont été observées chez 2,7 % (13/489) des patients. La colite d'origine immunologique a été de grade 3 chez 9 (1,8 %) de ces patients.
Le délai médian d'apparition de la colite d'origine immunologique était de 5,1 mois (intervalle : 2,3 semaines à 14 mois). La durée médiane de la réaction était de 1,6 semaine (intervalle : 1 jour à plus de 9 mois).
L'administration d'avélumab a été interrompue chez 0,4 % (2/489) des patients en raison d'une colite d'origine immunologique. Les 13 patients ayant présenté une colite d'origine immunologique ont tous été traités par corticothérapie et 12 (92,3 %) des 13 patients ont reçu une corticothérapie à forte dose pendant une durée médiane de 2,3 semaines (intervalle : 5 jours à 4,6 mois). La colite d'origine immunologique était résolue chez 10 (76,9 %) des 13 patients à la date limite de recueil des données.
Pancréatite d'origine immunologique
Chez les patients traités par avélumab en monothérapie, des pancréatites d'origine immunologique ont été observées chez moins de 1 % (1/4 000) des patients dans les études cliniques avec différents types de tumeurs et chez 0,6 % (3/489) des patients ayant reçu avélumab en association avec axitinib, l'issue ayant été fatale pour 2 (0,4 %) de ces 3 patients.
Myocardite d'origine immunologique
Chez les patients traités par avélumab en monothérapie, des myocardites d'origine immunologique ont été observées chez moins de 1 % (5/4 000) des patients dans les études cliniques avec différents types de tumeurs et chez 0,6 % (3/489) des patients ayant reçu avélumab en association avec axitinib, l'issue ayant été fatale chez 2 (0,4 %) de ces 3 patients.
Endocrinopathies d'origine immunologique Troubles thyroïdiens
Chez les patients traités par avélumab en monothérapie, 6 % (98/1 738) des patients ont présenté des troubles thyroïdiens d'origine immunologique, incluant 90 patients (5 %) avec une hypothyroïdie, 7 patients (0,4 %) avec une hyperthyroïdie et 4 patients (0,2 %) avec une thyroïdite. Les troubles thyroïdiens d'origine immunologique ont été de grade 3 chez 3 patients (0,2 %).
Le délai médian d'apparition des troubles thyroïdiens était de 2,8 mois (intervalle : 2 semaines à 13 mois). La durée médiane de la réaction n'a pu être évaluée (intervalle : 1 jour à plus de 26 mois).
L'administration d'avélumab a été interrompue chez 0,1 % (2/1 738) des patients en raison de troubles thyroïdiens d'origine immunologique. Les troubles thyroïdiens étaient résolus chez 7 (7 %) des 98 patients à la date limite de recueil des données.
Chez les patients traités par avélumab en association avec axitinib, des troubles thyroïdiens d'origine immunologique ont été observés chez 24,7 % (121/489) des patients, incluant 111 patients (22,7 %) avec une 'hypothyroïdie, 17 patients (3,5 %) avec une hyperthyroïdie et 7 patients (1,4 %) avec une thyroïdite. Les troubles thyroïdiens d'origine immunologique ont été de grade 3 chez 2 (0,4%) de ces patients.
Le délai médian d'apparition des troubles thyroïdiens était de 2,8 mois (intervalle : 3,6 semaines à 19,3 mois). La durée médiane de la réaction n'a pas pu être évaluée (intervalle : 8 jours à plus de 23,9 mois).
L'administration d'avélumab a été interrompue chez 0,2 % (1/489) des patients en raison de troubles thyroïdiens d'origine immunologique. Les troubles thyroïdiens étaient résolus chez 15 (12,4 %) des 121 patients à la date limite de recueil des données.
Insuffisance surrénalienne
Chez les patients traités par avélumab en monothérapie, une insuffisance surrénalienne d'origine immunologique a été observée chez 0,5 % (8/1 738) des patients. L'insuffisance surrénalienne d'origine immunologique a été de grade 3 chez 1 patient (0,1 %).
Le délai médian d'apparition de l'insuffisance surrénalienne d'origine immunologique était de 2,5 mois (intervalle : 1 jour à 8 mois). La durée médiane de la réaction n'a pu être évaluée (intervalle : 2 jours à plus de 6 mois).
L'administration d'avélumab a été interrompue chez 0,1 % (2/1 738) des patients en raison d'une insuffisance surrénalienne d'origine immunologique. Les 8 patients ayant présenté une insuffisance surrénalienne d'origine immunologique ont tous été traités par corticothérapie, et 4 (50 %) des 8 patients ont reçu de fortes doses de corticoïdes à action systémique (≥ 40 mg de prednisone ou équivalent) suivies d'une diminution progressive sur une durée médiane de 1 jour (intervalle : 1 jour à 24 jours). L'insuffisance surrénalienne était résolue chez 1 patient sous corticothérapie à la date limite de recueil des données.
Chez les patients traités par avélumab en association avec axitinib, une insuffisance surrénalienne d'origine immunologique a été observée chez 1,8 % (9/489) des patients. L'insuffisance surrénalienne d'origine immunologique a été de grade 3 chez 2 (0,4 %) de ces patients.
Le délai médian d'apparition de l'insuffisance surrénalienne d'origine immunologique était de 5,5 mois (intervalle : 3,6 semaines à 8,7 mois). La durée médiane de la réaction était de 2,8 mois (intervalle : 3 jours à 15,5 mois).
L'administration d'avélumab n'a été interrompue chez aucun patient en raison d'une insuffisance surrénalienne d'origine immunologique. Huit patients (88,9 %) ayant présenté une insuffisance surrénalienne d'origine immunologique ont été traités par corticothérapie et 2 (25 %) des 8 patients ont reçu de fortes doses de corticoïdes (≥ 40 mg de prednisone ou équivalent) pendant une durée médiane de 8 jours (intervalle : 5 jours à 11 jours). L'insuffisance surrénalienne était résolue chez 4 (44,4 %) des 9 patients à la date limite de recueil des données.
Diabète de type 1
Chez les patients traités par avélumab en monothérapie, des cas de diabète de type 1 sans autre étiologie connue ont été observés chez 0,1 % (2/1 738) des patients ; les deux réactions étaient de grade 3 et ont entraîné l'arrêt définitif d'avélumab.
Chez les patients traités par avélumab en association avec axitinib, un diabète de type 1 sans autre étiologie connue a été observé chez 1,0 % (5/489) des patients. Le diabète de type 1 était de grade 3 chez 1 (0,2 %) de ces patients.
Le délai médian d'apparition du diabète de type 1 était de 1,9 mois (intervalle : 1,1 mois à 7,3 mois).
L'administration d'avélumab a été interrompue chez 0,2 % (1/489) des patients en raison d'un diabète de type 1. Les 5 patients ayant présenté un diabète de type 1 ont tous été traités par insuline. Le diabète de type 1 n'était résolu chez aucun des patients au moment de la date limite de recueil des données.
Néphrite et dysfonction rénale d'origine immunologique
Chez les patients traités par avélumab en monothérapie, des cas de néphrite d'origine immunologique ont été observés chez 0,1 % (1/1 738) des patients traités par avélumab, entraînant l'arrêt définitif d'avélumab.
Chez les patients traités par avélumab en association avec axitinib, une néphrite d'origine immunologique a été observée chez 0,4 % (2/489) des patients. La néphrite d'origine immunologique était de grade 3 chez 2 (0,4 %) de ces patients.
Le délai médian d'apparition de la néphrite d'origine immunologique était de 1,2 mois (intervalle : 2,9 semaines à 1,8 mois). La durée médiane de la réaction était de 1,3 semaine (intervalle : plus de 4 jours à 1,3 semaine).
L'administration d'avélumab n'a été interrompue chez aucun patient en raison d'une néphrite d'origine immunologique. Les 2 patients ayant présenté une néphrite d'origine immunologique ont tous les deux été traités par de fortes doses de corticoïdes pendant une durée médiane de 1,1 semaine (intervalle : 3 jours à 1,9 semaine). La néphrite d'origine immunologique était résolue chez 1 (50 %) des 2 patients au moment de la date limite de recueil des données.
Hépatotoxicité (en association avec axitinib)
Chez les patients traités par avélumab en association avec axitinib, une augmentation des taux d'ALAT et d'ASAT de grade 3 et de grade 4 a été rapportée chez, respectivement, 9 % et 7 % des patients.
Chez les patients ayant présenté un taux d'ALAT ≥ 3 fois la LNS (grades 2-4, n = 82), le taux d'ALAT a régressé à un grade 0-1 dans 92 % des cas.
Parmi les 73 patients traités de nouveau par avélumab (59 %) ou axitinib (85 %) en monothérapie, ou les deux (55 %), 66 % n'ont pas montré de nouvelle augmentation du taux d'ALAT ≥ 3 fois la LNS.
Immunogénicité
Sur les 1 738 patients traités par avélumab à 10 mg/kg en perfusion intraveineuse toutes les 2 semaines, 1 627 étaient évaluables pour la détection d'anticorps anti-médicament (AAM) apparus sous traitement et le résultat était positif pour 96 (5,9 %) d'entre eux. Chez les patients AAM positifs, il peut y avoir un risque accru de réactions liées à la perfusion (environ 40 % chez les patients toujours AAM positifs et 25 % chez les patients jamais AAM positifs). Une nouvelle méthode plus sensible avec une tolérance médicamenteuse améliorée a été utilisée dans l'étude B9991001 pour détecter les AAM apparus sous traitement chez les patients traités par avélumab en monothérapie. Sur les 344 patients traités par avélumab à 10 mg/kg en perfusion intraveineuse toutes les 2 semaines associé aux meilleurs soins de soutien (BSC, best supportive care), 325 étaient évaluables pour la détection des AAM apparus sous traitement et le résultat était positif pour 62 (19,1 %) d'entre eux. D'après les données disponibles, notamment la faible incidence de l'immunogénicité, l'impact des AAM sur la pharmacocinétique, l'efficacité et la sécurité du médicament est incertain. L'impact sur les anticorps neutralisants (AcN) n'est pas connu.
Sur les 480 patients ayant été traités par avélumab à 10 mg/kg en perfusion intraveineuse toutes les 2 semaines en association avec 5 mg d'axitinib deux fois par jour et pour lesquels au moins une mesure valide des AAM était disponible à un moment quelconque, 453 étaient évaluables pour la détection d'AAM apparus sous traitement et le résultat était positif pour 66 (14,6 %) d'entre eux. La nouvelle méthode de détection des AAM, dotée d'une sensibilité améliorée, a été utilisée dans la population atteinte de CCR. Globalement, aucune modification du profil pharmacocinétique, aucune augmentation de l'incidence des réactions à la perfusion ni d'effets sur l'efficacité en lien avec l'apparition d'anticorps anti-avélumab n'ont été mis en évidence.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : signalement.social-sante.gouv.fr.
AVANT LE TRAITEMENT :
Les patients doivent recevoir une prémédication composée d'un
antihistaminique et de paracétamol avant les 4 premières perfusions. Si
la quatrième perfusion a pu être réalisée sans que survienne de
réaction liée à la perfusion, l'administration de la prémédication
avant les doses suivantes sera laissée à l'appréciation du médecin.
SURVEILLANCE PENDANT LE TRAITEMENT :
- Signes et symptômes de réactions liées à la perfusion, notamment
fièvre, frissons, bouffées vasomotrices, hypotension, dyspnée,
sifflement respiratoire, dorsalgie, douleur abdominale et urticaire.
- Signes et symptômes de pneumopathie d'origine immunologique.
- Signes et symptômes de colite d'origine immunologique.
- Signes et symptômes de pancréatite d'origine immunologique.
- Signes et symptômes de myocardite d'origine immunologique.
- Signes et symptômes cliniques d'endocrinopathies.
- Signes et symptômes d'altération de la fonction hépatique.
- Les altérations de la fonction thyroïdienne doivent être surveillées
chez les patients (en début de traitement, régulièrement au cours du
traitement et dès lors qu'un suivi est indiqué en fonction de
l'évaluation clinique), ainsi que les signes cliniques et les symptômes
de troubles thyroïdiens.
- Les signes et symptômes d'insuffisance surrénalienne doivent être
surveillés chez les patients pendant et après le traitement.
- Les patients doivent être surveillés pour rechercher une hyperglycémie ou d'autres signes et symptômes du diabète.
- Les patients doivent être surveillés pour rechercher des taux
sériques élevés de créatinine avant le traitement et, à intervalles
réguliers, pendant le traitement.
Femmes en âge de procréer/contraception
Il doit être conseillé aux femmes en âge de procréer d'éviter de débuter une grossesse pendant qu'elles reçoivent avélumab. Elles doivent utiliser une contraception efficace pendant le traitement par avélumab et pendant au moins 1 mois après la dernière dose d'avélumab.
Grossesse
Il n'existe pas de données ou il existe des données limitées sur l'utilisation d'avélumab chez la femme enceinte.
Aucune étude des effets d'avélumab sur la reproduction animale n'a été effectuée. Cependant, il a été démontré chez les modèles murins gravides que le blocage de la voie de signalisation PD-L1 entraînait une perturbation de la tolérance foeto-maternelle, aboutissant à une augmentation des pertes foetales (voir rubrique Données de sécurité précliniques). Ces résultats indiquent que l'administration d'avélumab pendant la grossesse, étant donné son mécanisme d'action, expose à un risque potentiel d'effets délétères sur le foetus, notamment à des taux plus élevés d'avortement ou de mort-né.
Les immunoglobulines IgG1 humaines sont connues pour traverser la barrière placentaire. Avélumab est donc susceptible de passer de la mère au foetus en cours de développement. Il n'est pas recommandé d'utiliser avélumab pendant la grossesse à moins que la situation clinique de la femme ne justifie le traitement par avélumab.
Allaitement
On ne sait pas si avélumab est excrété dans le lait maternel. Dans la mesure où l'on sait que les anticorps peuvent être sécrétés dans le lait maternel, un risque pour les nouveau-nés/nourrissons ne peut être exclu.
Il doit être conseillé aux femmes de ne pas allaiter pendant le traitement et pendant au moins 1 mois après la dernière dose en raison du risque potentiel d'effets indésirables graves chez les nourrissons allaités.
Fertilité
Les effets d'avélumab sur la fertilité masculine et féminine ne sont pas connus.
Bien qu'aucune étude n'ait été réalisée pour évaluer les effets d'avélumab sur la fertilité, il n'a été constaté aucun effet notable sur les organes reproducteurs des singes femelles lors des études de toxicologie en administration répétée sur 1 mois et 3 mois (voir rubrique Données de sécurité précliniques).
Aucune étude d'interaction n'a été réalisée avec avélumab.
Avélumab étant principalement métabolisé par des voies du catabolisme, aucune interaction pharmacocinétique entre avélumab et d'autres médicaments n'est attendue.
Le traitement doit être instauré et supervisé par un médecin expérimenté en matière de traitement du cancer.
Posologie
La dose recommandée de Bavencio en monothérapie est de 800 mg à administrer par voie intraveineuse pendant 60 minutes toutes les 2 semaines.
L'administration de Bavencio doit être poursuivie selon le schéma recommandé jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable.
Prémédication
Les patients doivent recevoir une prémédication composée d'un antihistaminique et de paracétamol avant les 4 premières perfusions de Bavencio. Si la quatrième perfusion a pu être réalisée sans que survienne de réaction liée à la perfusion, l'administration de la prémédication avant les doses suivantes sera laissée à l'appréciation du médecin.
Modifications du traitement
Les augmentations ou réductions de dose ne sont pas recommandées. Un report ou une interruption du traitement peuvent être nécessaires au cas par cas en fonction de la sécurité d'emploi et de la tolérance ; voir le Tableau 1.
Les recommandations précises pour la prise en charge des effets indésirables d'origine immunologique sont présentées dans la rubrique Mises en garde et précautions d'emploi.
Tableau 1 : Recommandations pour la suspension ou l'arrêt de Bavencio
| Effet indésirable lié au traitement | Sévérité* | Modification du traitement |
| Réaction liée à la perfusion | Réaction liée à la perfusion de grade 1 | Réduire le débit de perfusion de 50 % |
| Réaction liée à la perfusion de grade 2 | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 ; reprendre la perfusion à un débit réduit de 50 % | |
| Réaction liée à la perfusion de grade 3 ou de grade 4 | Arrêter définitivement le traitement | |
| Pneumopathie | Pneumopathie de grade 2 | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 |
| Pneumopathie de grade 3 ou de grade 4, ou pneumopathie récurrente de grade 2 | Arrêter définitivement le traitement | |
| Hépatite | Aspartate aminotransférases (ASAT) ou alanine aminotransférases (ALAT) supérieures à 3 fois et inférieures ou égales à 5 fois la limite normale supérieure (LNS) ou bilirubine totale supérieure à 1,5 fois et inférieure ou égale à 3 fois la LNS | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 |
| ASAT ou ALAT supérieures à 5 fois la LNS ou bilirubine totale supérieure à 3 fois la LNS | Arrêter définitivement le traitement | |
| Colite | Colite ou diarrhée de grade 2 ou de grade 3 | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 |
| Colite ou diarrhée de grade 4 ou colite récurrente de grade 3 | Arrêter définitivement le traitement | |
| Pancréatite | Pancréatite suspectée | Suspendre le traitement |
| Pancréatite confirmée | Arrêter définitivement le traitement | |
| Myocardite | Myocardite suspectée | Suspendre le traitement |
| Myocardite confirmée | Arrêter définitivement le traitement | |
| Endocrinopathies (hypothyroïdie, hyperthyroïdie, insuffisance surrénalienne, hyperglycémie) | Endocrinopathies de grade 3 ou de grade 4 | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 |
| Néphrite et dysfonction rénale | Taux de créatinine sérique compris entre 1,5 fois et 6 fois la LNS | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 |
| Taux de créatinine sérique supérieur à 6 fois la LNS | Arrêter définitivement le traitement |
| Effet indésirable lié au traitement | Sévérité* | Modification du traitement |
| Autres effets indésirables d'origine immunologique (dont myosite, hypopituitarisme, uvéite, syndrome de Guillain-Barré) | Pour les effets suivants : · Signes ou symptômes cliniques de grade 2 ou de grade 3 d'un effet indésirable d'origine immunologique non décrit ci-dessus. | Suspendre le traitement jusqu'à régression des effets indésirables à un grade 0-1 |
| Pour les effets suivants : · Effet indésirable de grade 4 ou engageant le pronostic vital (à l'exception des endocrinopathies contrôlées par un traitement hormonal substitutif) · Effet indésirable d'origine immunologique récurrent de grade 3 · Nécessité d'administrer 10 mg/jour ou plus de prednisone ou équivalent pendant plus de 12 semaines · Effets indésirables à médiation immunitaire de grade 2 ou de grade 3 persistants pendant 12 semaines ou plus | Arrêter définitivement le traitement |
* Toxicité évaluée selon les Critères Communs de Terminologie pour les Effets Indésirables du National Cancer Institute, version 4.0 (NCI-CTCAE v4.03)
Populations particulières Personnes âgées
Aucun ajustement posologique n'est nécessaire chez les patients âgés (≥ 65 ans) (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de Bavencio chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été établies.
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale légère ou modérée (voir rubrique Propriétés pharmacocinétiques). Les données concernant les patients atteints d'insuffisance rénale sévère sont insuffisantes pour donner des recommandations posologiques.
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique légère (voir rubrique Propriétés pharmacocinétiques). Les données concernant les patients atteints d'insuffisance hépatique modérée ou sévère sont insuffisantes pour donner des recommandations posologiques.
Mode d'administration
Bavencio est destiné à être utilisé en perfusion intraveineuse exclusivement. Il ne doit pas être administré en injection intraveineuse rapide ni en bolus intraveineux.
Bavencio doit être dilué dans une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou une solution injectable de chlorure de sodium à 4,5 mg/mL (0,45 %). Il sera ensuite administré en perfusion intraveineuse pendant 60 minutes en utilisant un filtre intégré ou additionnel stérile, apyrogène, à faible liaison aux protéines, de 0,2 micromètre.
Pour les instructions concernant la préparation et l'administration du médicament, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
Flacon non ouvert 2 ans
Après ouverture
D'un point de vue microbiologique, après ouverture, le médicament doit être dilué et perfusé immédiatement.
Après préparation de la solution pour perfusion
La stabilité chimique et physique de la solution diluée a été démontrée sur une durée de 24 heures à une température comprise entre 20°C et 25°C, et sous éclairage intérieur. D'un point de vue microbiologique, à moins que la méthode de dilution utilisée ne permette d'exclure tout risque de contamination microbienne, la solution diluée doit être perfusée immédiatement. Si la solution n'est pas utilisée immédiatement, la durée et les conditions de conservation avant utilisation sont de la responsabilité de l'utilisateur.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
À conserver dans l'emballage extérieur d'origine à l'abri de la lumière.
Pour les conditions de conservation du médicament après dilution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Bavencio doit être dilué dans une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou une solution injectable de chlorure de sodium à 4,5 mg/mL (0,45 %).
Un surdosage a été rapporté chez trois patients qui ont reçu une dose d'avélumab entre 5 % à 10 % supérieure à la dose recommandée. Les patients n'ont présenté aucun symptôme, le surdosage n'a nécessité aucun traitement et l'administration d'avélumab a été poursuivie.
En cas de surdosage, les signes ou symptômes d'effets indésirables doivent être étroitement surveillés chez les patients. Le traitement visera à prendre en charge les symptômes.
Classe pharmacothérapeutique : autres agents antinéoplasiques, anticorps monoclonaux, code ATC : L01XC31.
Mécanisme d'action
Avélumab est un anticorps monoclonal humain de type immunoglobuline G1 (IgG1) dirigé contre le ligand de la protéine de mort programmée 1 (PD-L1). Avélumab se lie au PD-L1 et bloque l'interaction entre le PD-L1 et ses récepteurs, PD-1 (protéine de mort programmée 1) et B7.1. Cela conduit à la suppression des effets inhibiteurs du PD-L1 sur les lymphocytes T CD8+ cytotoxiques, rétablissant ainsi les réponses anti-tumorales des lymphocytes T.
Il a également été montré qu'avélumab induisait une lyse directe des cellules tumorales par les cellules Natural Killer (NK) via la cytotoxicité à médiation cellulaire dépendante des anticorps (CCDA).
Efficacité et sécurité clinique
Carcinome urothélial localement avancé ou métastatique (étude B9991001)
L'efficacité et la sécurité d'avélumab ont été évaluées dans le cadre de l'étude B9991001, une étude de phase III randomisée, multicentrique, en ouvert, réalisée chez 700 patients atteints d'un carcinome urothélial localement avancé non résécable ou métastatique, dont la maladie n'avait pas progressé après une chimiothérapie d'induction de première ligne à base de sels de platine. Les patients présentant une maladie auto-immune ou une affection nécessitant un traitement immunosuppresseur étaient exclus.
La randomisation a été stratifiée selon la meilleure réponse à la chimiothérapie (RC/RP vs. maladie stable [MS]) et le site des métastases (viscérales vs. non-viscérales) au moment de l'instauration de la chimiothérapie d'induction de première ligne. Les patients ont été randomisés (1:1) pour recevoir avélumab à 10 mg/kg en perfusion intraveineuse toutes les 2 semaines associé aux meilleurs soins de soutien (BSC, best supportive care) ou meilleurs soins de soutien uniquement.
Le traitement par avélumab a été poursuivi jusqu'à la progression de la maladie telle que définie par les critères d'évaluation des réponses tumorales relatives aux tumeurs solides (RECIST, Response Evaluation Criteria in Solid Tumours v 1.1) et constatée par un examen centralisé indépendant réalisé en aveugle (BICR, Blinded Independent Central Review), ou jusqu'à l'apparition d'une toxicité inacceptable. L'administration d'avélumab était permise au-delà de la progression de la maladie définie par les critères RECIST si le patient était cliniquement stable et si l'investigateur estimait que le patient en retirait un bénéfice clinique. L'évaluation du statut tumoral a été réalisée à l'inclusion, 8 semaines après la randomisation, puis toutes les 8 semaines jusqu'à 12 mois après la randomisation, puis toutes les 12 semaines jusqu'à confirmation de la progression de la maladie basée sur le BICR d'après les critères RECIST v 1.1.
Les caractéristiques démographiques à l'inclusion étaient généralement bien équilibrées entre le bras avélumab plus BSC et le bras BSC seul. Les caractéristiques à l'inclusion étaient les suivantes : âge médian de 69 ans (min : 32 ans, max : 90 ans), 66 % des patients étaient âgés de 65 ans ou plus, 77 % étaient de sexe masculin, 67 % étaient caucasiens et l'indice de performance ECOG était de 0 (61 %) ou 1 (39 %) pour les deux bras.
Pour la chimiothérapie d'induction de première ligne, 56 % des patients ont reçu du cisplatine associé à la gemcitabine, 38 % des patients du carboplatine associé à la gemcitabine, et 6 % des patients du cisplatine associé à la gemcitabine et du carboplatine associé à la gemcitabine (c'est-à-dire ces patients ont reçu un ou plusieurs cycles de chaque association). La meilleure réponse à la chimiothérapie d'induction de première ligne était RC ou RP (72 %) ou MS (28 %). Les sites des métastases avant la chimiothérapie étaient des métastases viscérales (55 %) ou non-viscérales (45 %). Cinquante et un pourcent des patients avaient des tumeurs PD-L1 positives. Six pourcent des patients dans le bras avélumab plus BSC et 44 % des patients dans le bras BSC seul ont reçu un autre inhibiteur de checkpoint PD-1/PD-L1 après arrêt du traitement.
Le critère d'efficacité primaire était la survie globale (SG) chez tous les patients randomisés et les patients ayant des tumeurs PD-L1-positives. La survie sans progression (SSP) basée sur le BICR d'après les critères RECIST v 1.1 était un critère d'efficacité secondaire. Les résultats d'efficacité ont été mesurés à partir du moment de la randomisation c'est-à-dire après 4 à 6 cycles de chimiothérapie d'induction à base de sels de platine.
Les résultats d'efficacité sont présentés ci-dessous.
Tableau 4 : Résultat d'efficacité de l'étude B9991001
| Critères d'efficacité | Avélumab plus BSC (N=350) | BSC (N=350) |
| Survie globale (SG) | | |
| Evénements (%) | 145 (41,4) | 179 (51,1) |
| Médiane en mois (IC à 95 %) | 21,4 (18,9, 26,1) | 14,3 (12,9, 17,9) |
| Rapport de risque (IC à 95 %) | 0,69 (0,556, 0,863) | |
| Valeur de p (test bilatéral)* | 0,0010 | |
| Taux de SG à 12 mois selon la méthode K-M (IC à 95 %)** | 71,3 % (66,0, 76,0) | 58,4 % (52,7, 63,7) |
| Taux de SG à 18 mois selon la méthode K-M (IC à 95 %)** | 61,3 % (55,4, 66,7) | 43.8 % (37,8, 49,7) |
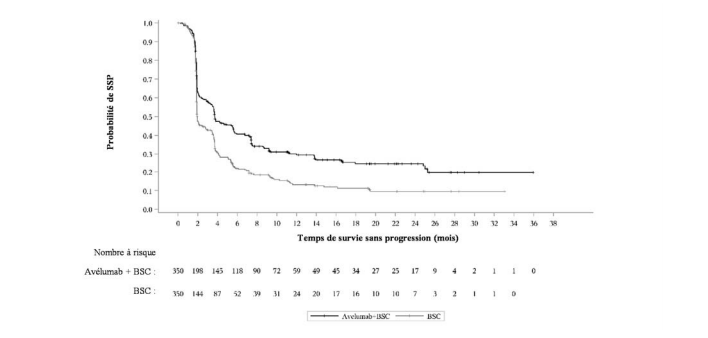
| Survie sans progression (SSP)*** | | |
| Evénements (%) | 225 (64,3) | 260 (74,3) |
| Médiane en mois (IC à 95 %) | 3,7 (3,5, 5,5) | 2,0 (1,9, 2,7) |
| Rapport de risque (IC à 95 %) | 0,62 (0,519, 0,751) | |
| Valeur de p (test bilatéral)* | < 0,0001 | |
IC : intervalle de confiance ; K-M : Kaplan-Meier
* Valeur de p basée sur le log-rank stratifié
** Les IC sont calculés à l'aide de la transformation log-log avec rétro-transformation en une échelle non- transformée
*** Basée sur le BICR d'après les critères RECIST v 1.1
Figure 1 : Estimations de Kaplan-Meier de la survie globale (SG) - Echantillon complet d'analyse
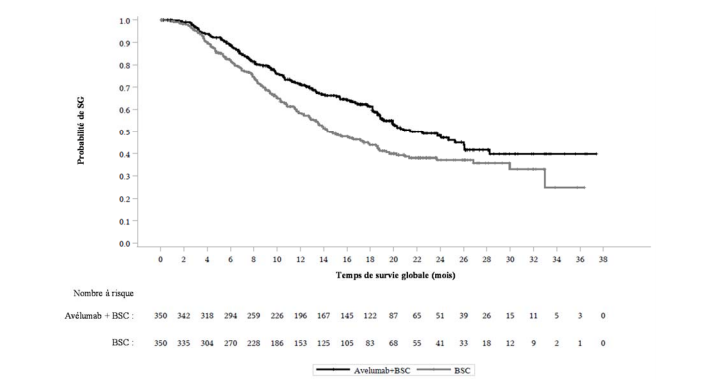
Figure 2 : Estimations de Kaplan-Meier de la survie sans progression (SSP) basée sur le BICR d'après les critères RECIST v 1.1 - Echantillon complet d'analyse
De plus, une amélioration statistiquement significative de la SG a été démontrée chez les patients avec des tumeurs PD-L1 positives pour l'association avélumab plus BSC par rapport à BSC seul (HR 0,56 ; IC à 95 % : 0,40 ; 0,79 ; valeur de p (test bilatéral) 0,0007). La SG médiane n'a pas été atteinte (IC à 95% : 20,3 mois, non atteinte) dans le bras avélumab plus BSC, et était de 17,1 mois (IC à 95% : 13,5, 23,7) dans le bras BSC seul.
Des résultats cohérents ont été observés dans les sous-groupes établis, notamment pour la meilleure réponse à la chimiothérapie d'induction de première ligne, les sites des métastases et le statut d'expression tumorale de PD-L1 comme le montre la figure 3.
Figure 3 : Graphique en forêt de la survie globale (SG) par sous-groupe - Echantillon complet d'analyse
Résultats rapportés par les patients (PRO, Patient Reported Outcomes)
Les résultats rapportés par les patients en termes de symptômes physiques et émotionnels liés à la maladie, effets secondaires du traitement, fonctions et bien-être ont été collectés à l'aide du questionnaire FACT/NCCN Bladder Symptom Index (FBlSI-18). Aucun effet néfaste n'a été observé lors de l'ajout d'un traitement d'entretien par avélumab à BSC par rapport à BSC seul selon le questionnaire FBlSI-18 pendant la période de traitement.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Bavencio dans tous les sous-groupes de la population pédiatrique pour le traitement du carcinome urothélial (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La pharmacocinétique (PK) d'avélumab a été évaluée suivant une approche PK de population.
D'après l'analyse PK de population, aucune différence cliniquement significative n'est attendue au niveau de l'exposition à avélumab entre les schémas d'administration toutes les 2 semaines à 800 mg ou à 10 mg/kg.
Distribution
Une distribution d'avélumab dans la circulation générale est escomptée et, dans une moindre mesure, dans le compartiment extracellulaire. Le volume de distribution à l'état d'équilibre est de 4,72 L.
En cohérence avec la distribution extravasculaire limitée, le volume de distribution d'avélumab à l'état d'équilibre est faible. Comme attendu pour un anticorps, avélumab ne se lie pas de façon spécifique aux protéines plasmatiques.
Élimination
D'après l'analyse pharmacocinétique de population effectuée chez 1 629 patients, la clairance systémique totale (CL) est de 0,59 L/jour. Lors de l'analyse complémentaire, il a été observé que la CL d'avélumab diminuait avec le temps : la réduction maximale moyenne la plus importante (coefficient de variation [CV%]) par rapport à la valeur initiale avec différents types de tumeurs a été d'environ 32,1 % (CV de 36,2 %).
Les concentrations d'avélumab ont atteint l'état d'équilibre au bout d'environ 4 à 6 semaines (2 à 3 cycles) d'administration répétée à 10 mg/kg toutes les 2 semaines et une accumulation systémique d'un facteur 1,25 environ a été observée.
À la dose recommandée, la demi-vie d'élimination (t½) est de 6,1 jours, d'après l'analyse PK de population.
Linéarité/non-linéarité
L'exposition à avélumab augmente de façon proportionnelle à la dose dans l'intervalle posologique de 10 mg/kg à 20 mg/kg toutes les 2 semaines.
Populations particulières
L'analyse pharmacocinétique de population n'a révélé aucune différence au niveau de la clairance systémique totale d'avélumab en fonction de l'âge, du sexe, de l'origine ethnique, du statut PD-L1, de la charge tumorale, de la présence d'une insuffisance rénale ou d'une insuffisance hépatique légère ou modérée.
La clairance systémique totale augmente avec le poids corporel. L'exposition à l'état d'équilibre était approximativement uniforme sur un large intervalle de poids corporels (30 à 204 kg) pour une dose normalisée au poids corporel.
Insuffisance rénale
Aucune différence cliniquement notable n'a été constatée au niveau de la clairance d'avélumab entre les patients présentant une insuffisance rénale légère (débit de filtration glomérulaire [DFG] de 60 à 89 mL/min, clairance de la créatinine [ClCr] selon la formule de Cockroft-Gault ; n = 623), modérée (DFG de 30 à 59 mL/min, n = 320) et les patients présentant une fonction rénale normale (DFG
≥ 90 mL/min, n = 671).
Avélumab n'a pas été évalué chez les patients atteints d'insuffisance rénale sévère (DFG de 15 à 29 mL/min).
Insuffisance hépatique
Aucune différence cliniquement notable n'a été constatée au niveau de la clairance d'avélumab entre les patients présentant une insuffisance hépatique légère (bilirubine ≤ LNS et ASAT > LNS ou bilirubine comprise entre 1 et 1,5 fois la LNS, n = 217) et ceux présentant une fonction hépatique normale (bilirubine et ASAT ≤ LNS, n = 1 388) lors de l'analyse PK de population. L'insuffisance hépatique était définie selon les critères du National Cancer Institute (NCI) pour l'évaluation de la dysfonction hépatique.
Avélumab n'a pas été évalué chez les patients atteints d'insuffisance hépatique modérée (bilirubine comprise entre 1,5 et 3 fois la LNS) ou d'insuffisance hépatique sévère (bilirubine > 3 fois la LNS).
Avélumab a un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Des cas de fatigue ont été signalés après l'administration d'avélumab (voir rubrique Effets indésirables). Il doit être conseillé aux patients d'être prudents lorsqu'ils conduisent des véhicules ou utilisent des machines tant qu'ils ne sont pas certains que le traitement par avélumab n'altère pas leur vigilance.
Les données non cliniques issues des études conventionnelles de toxicologie en administration répétée chez des singes cynomolgus traités par voie intraveineuse à des doses de 20, 60 ou 140 mg/kg une fois par semaine pendant 1 mois et 3 mois, avec une période de récupération de 2 mois après la période d'administration de 3 mois, n'ont pas révélé de risque particulier pour l'homme. Une infiltration périvasculaire de cellules mononucléées a été observée dans le cerveau et la moelle épinière des singes traités par avélumab à une dose ≥ 20 mg/kg pendant 3 mois. Bien qu'aucune relation dose- réponse claire n'ait été mise en évidence, un lien entre cette observation et le traitement par avélumab ne peut être exclu.
Aucune étude des effets d'avélumab sur la reproduction animale n'a été effectuée. La voie PD-1/PD-L1 pourrait être impliquée dans le maintien de la tolérance au foetus tout au long de la grossesse. Il a été montré, chez les modèles murins gravides, que le blocage de la voie de signalisation du PD-L1 perturbait la tolérance au foetus, entraînant une augmentation des pertes foetales. Ces résultats indiquent que l'administration d'avélumab pendant la grossesse expose à un risque potentiel d'effets délétères sur le foetus, notamment à des taux plus élevés d'avortement ou de mort-né.
Aucune étude n'a été réalisée pour évaluer le potentiel cancérogène ou génotoxique d'avélumab.
Les effets d'avélumab sur la fertilité n'ont pas été étudiés. Les études de toxicologie en administration répétée sur 1 mois et 3 mois chez le singe n'ont révélé aucun effet notable sur les organes reproducteurs femelles. Un grand nombre des singes mâles utilisés au cours de ces études étaient sexuellement immatures ; par conséquent, aucune conclusion explicite ne peut être tirée concernant les effets sur les organes reproducteurs mâles.
Bavencio est compatible avec les poches de perfusion en polyéthylène, en polypropylène et en éthylène-acétate de vinyle, avec les flacons en verre, le matériel de perfusion en polychlorure de vinyle et les filtres intégrés avec membranes en polyéther sulfone dotées de pores d'un diamètre de 0,2 micromètre.
Instructions de manipulation
La solution pour perfusion doit être préparée en respectant les règles d'asepsie.
· Le flacon doit être inspecté visuellement afin de rechercher les possibles particules ou changements de coloration. Bavencio est une solution limpide, incolore à légèrement jaune. Si la solution est trouble, a changé de couleur ou contient des particules, le flacon doit être éliminé.
· Une poche de perfusion de dimension appropriée (de préférence 250 mL) contenant une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou une solution injectable de chlorure de sodium à 4,5 mg/mL (0,45 %) doit être utilisée. Le volume nécessaire de Bavencio doit être prélevé dans le(s) flacon(s) et transféré dans la poche de perfusion. Tout flacon entamé ou vide doit être éliminé.
· La solution diluée doit être mélangée en retournant délicatement la poche de façon à éviter de faire mousser la solution ou de la soumettre à une force de cisaillement trop importante.
· La solution doit être inspectée afin de vérifier qu'elle est limpide, incolore et exempte de particules visibles. Une fois préparée, la solution diluée doit être utilisée immédiatement.
· Les autres médicaments ne doivent pas être coadministrés dans la même ligne de perfusion intraveineuse. La perfusion doit être administrée en utilisant un filtre intégré à la ligne de perfusion ou additionnel stérile, apyrogène, à faible liaison aux protéines, de 0,2 micromètre comme indiqué dans la rubrique Posologie et mode d'administration.
Après l'administration de Bavencio, la ligne de perfusion doit être purgée à l'aide d'une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou d'une solution injectable de chlorure de sodium à 4,5 mg/mL (0,45 %).
Ne pas congeler ni agiter la solution diluée. En cas de réfrigération, laisser la solution diluée dans les poches de perfusion intraveineuse revenir à température ambiante avant utilisation.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament réservé à l'usage hospitalier.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancéro.
Médicament nécessitant une surveillance particulière pendant le traitement.
Solution à diluer pour perfusion (solution à diluer stérile).
Solution limpide, incolore à légèrement jaune.
10 mL de solution à diluer dans un flacon (en verre de type I) fermé par un bouchon en caoutchouc halo butyle et un opercule scellé en aluminium avec capuchon amovible en plastique.
Boîte de 1 flacon.